

环肽-类肽杂合物比线性肽具有更高的稳定性和选择性,因此是更好的药物候选物。然而,它们的合成远非易事,通常难以实现自动化。在此,我们描述了一种基于点击化学的环肽-类肽杂合物的快速高效合成新方法。我们的方法基于易于合成的构建模块、自动化微波辅助固相合成以及生物正交点击环化反应的结合。我们使用先前已证明可激活 HIV-1 整合酶的 INS 肽来验证此方法的概念。该策略能够以高产率和高纯度快速合成并进行生物物理评估源自 HIV-1 整合酶的环肽-类肽杂合物库。新的环杂合物显示出增强的生物活性,并且比原始的线性 INS 肽稳定得多。
简介:线性肽作为药物先导化合物存在若干缺点,例如由于酶促降解、水解或氧化导致的代谢不稳定,半衰期短、清除率快,口服生物利用度低,在某些情况下溶解度差和膜通透性低。肽环化是一种克服这些缺点的有效策略。环化是从自然界借鉴的一种方法,用于限制肽的构象,从而提高肽对靶蛋白的亲和力和选择性。环肽可分为头尾环化、头侧链环化、侧链尾环化和侧链侧链环化。已采用多种方法生成环肽,如主链环化、肽钉合和天然化学连接。这些结构可以通过化学稳定的键形成,例如酰胺键、内酯键、醚键、硫醚键或二硫键。肽环化可以通过使用天然氨基酸侧链实现,包括半胱氨酸二硫键、胺芳基化和烷基化等。此外,包括非天然氨基酸在内的反应也常被使用,例如环合复分解反应、点击化学等等。环肽受限的构象使其更不易降解和发生化学修饰,从而增强了环肽的稳定性。进入临床试验或已获批的环肽数量大幅增加。例如,环孢素 A 由于其免疫抑制活性,自 2001 年获得美国食品药品监督管理局(FDA)批准以来,一直用于治疗同种异体移植物。罗米地辛于 2009 年获 FDA 批准用于皮肤 T 细胞淋巴瘤,2011 年获批用于外周 T 细胞淋巴瘤。
另一种改善肽类药物性质的方法是通过引入肽键模拟物将肽转化为肽模拟物。例如,在具有生物活性的肽中加入 D 型氨基酸或非天然氨基酸可以提高其代谢稳定性。非天然氨基酸肌氨酸被用于开发 ,这是一种血管紧张素 II 类似物,用作血管紧张素 II 受体的部分激动剂,用于治疗高血压。用非天然氨基酸肌氨酸替代有助于抵抗氨肽酶降解,从而提高了化合物的生物活性。肽类模拟物是一类由 N-取代甘氨酸低聚物组成的化合物。与肽不同,肽类模拟物的侧链连接在主链氮原子上,而不是连接在手性α-碳原子上。肽类模拟物对蛋白水解高度稳定,并且能够折叠成明确的二级结构。
开发新的快速合成路线以将肽环化与肽键模拟物相结合的需求十分紧迫。将肽环化与肽素相结合能够显著改善先导肽的药理特性。例如,将类肽残基引入环肽中,其细胞渗透性优于全肽或全类肽序列。已有若干报告介绍了环肽 -类肽杂合物的合成。例如,一系列包含不同类肽侧链的环肽 -类肽支架通过平行人工膜测试了被动膜渗透性。这些变体无论其侧链上的官能团如何,都具有内在的渗透性。还展示了针对注射器 T3SS 的高选择性环肽 -类肽杂合物抑制剂,且不影响鞭毛 T3SS。环肽 -类肽杂合物还被用作细菌群体感应调节剂、黑皮质素受体激动剂和组蛋白去乙酰化酶抑制剂。这些分子在结构多样性和生物功能方面比简单的肽或肽类衍生物具有许多优势。因此,开发用于合成环肽 – 肽类衍生物杂合物的新且快速的合成方法也可用于高通量制备库。
铜(I)催化的叠氮化物与炔烃的[3+2] 环加成反应(CuAAC)是有机化学和药物发现领域中应用最广泛的点击反应之一。点击化学是一种高效的工具,可用于将肽与放射性分子或荧光化合物、其他生物活性分子(如用作药物的小分子或核酸)连接起来,也可用于肽环化。CuAAC 反应被广泛使用,因为该反应非常稳健、选择性强、对 pH 值和温度的变化不敏感,并且可以在天然官能团存在的情况下进行。微波辅助(MW)合成能够利用典型的点击反应方案促进三唑的形成并加快其生成速度。在肽和碳水化合物缀合物的制备中使用了微波辅助点击反应。
利用点击化学合成环肽 – 类肽杂合物可以通过两种不同的方法实现:亚单体法和构建模块法。亚单体法在多个案例中均有报道,包括两步程序:(i)将溴乙酸与树脂结合的正在生长的受保护肽链的 N 端游离胺偶联,(ii)用所需的胺进行烷基化(图1)。目前市面上有数百种伯胺可供使用,可通过亚单体法合成肽-类肽库。然而,现有的亚单体法方案需要多个合成步骤,每次烷基化都需要手动添加试剂,因此该策略无法完全实现自动化。此外,沸点低且碳链短的烷基胺与微波辅助合成方案不兼容,其中一些在市场上也难以买到。另一方面,构建块法通过将保护的 N-烷基化氨基酸与肽序列在一步中偶联来实现,就像任何其他标准保护氨基酸一样。这种方法能够以自动化的方式将肽素构建块(BBs)整合到标准固相肽合成(SPPS)方案中,并具有微波使用的优点。这种合成的主要困难在于制备具有合适保护基团的多样化 BBs,这些保护基团还应与微波-SPPS 条件兼容。
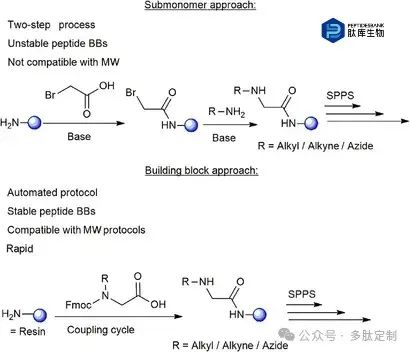
图 1. 用于合成环肽-肽素杂化物的亚单体和构建单元方法的图示。展示了每种方法的主要优点和局限性。树脂以蓝色球体表示。
为应对上述挑战,我们描述了一种新的快速合成方法的开发,该方法几乎完全自动化,可在数小时内提供所需的环肽-类肽杂合物。为实现这一目标,我们合成了叠氮基和炔基 N-烷基化氨基酸,利用固相肽合成法将其引入肽序列,并采用优化后的点击化学方案对肽进行环化,该方案专为微波固相肽合成设计。用于验证新环化方法概念的肽是源自 HIV-1 整合酶(IN)的肽 IN181−188。这是我们在实验室开发的 IN 激活肽 INS(源自 IN 氨基酸 174-188)的一个短片段,我们已证明其能通过诱导多整合和细胞凋亡杀死 HIV 感染细胞。丙氨酸扫描表明,截短肽 IN181−188 与 IN 蛋白的结合和激活效率与 INS 本身相当,因此用它代替了长的母肽。
结果
肽类-肽样化合物构建模块的合成
为了合成多样化的环肽-肽样化合物杂合体库,需要将相应的肽样化合物构建模块整合到肽序列中。设计并合成了三种不同链长的非天然Fmoc保护的肽样化合物构建模块。这些构建模块能够像常规氨基酸一样,通过标准的自动化微波Fmoc-SPPS程序轻松地插入到肽序列中的指定位置。通过开发新的合成策略制备了叠氮基和炔基肽样化合物构建模块(图2)。通过在二异丙基乙胺(DIEA)和干燥的四氢呋喃(THF)存在下使甘氨酸甲酯与丙炔基溴反应合成了丙炔基构建模块1a。还测试了其他几种碱,如三乙胺和吡啶,但DIEA能以最佳产率得到粗中间体1。通过薄层色谱(TLC)测定中间体的纯度为80-85%。在DIEA存在下,用氢氧化锂水解并用Fmoc保护胺基,得到所需的Fmoc保护的炔基构建模块1a(图2A)。粗制的 N-炔丙基甘氨酸 1a 经过与乙醚研磨,并从二氯甲烷(DCM)和己烷中重结晶,从而得到纯化合物。
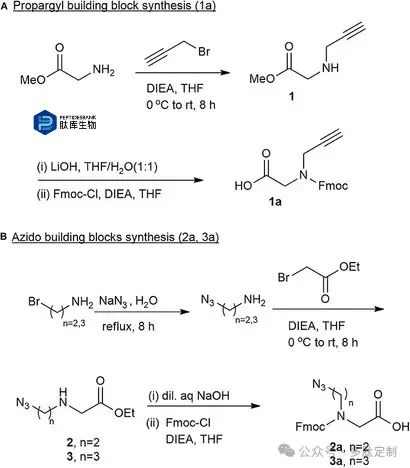
图 2. 本研究中合成并使用的 Fmoc 保护的肽素构建单元。 (A) 合成丙炔基构建单元 1a。 (B) 合成叠氮化物构建单元。 2a 在主链中含两个碳原子,3a 在主链中含三个碳原子。
两个N-叠氮基烷基甘氨酸构建模块是通过类似的方案合成的。在回流条件下,叠氮化钠处理2-溴乙胺和3-溴丙胺,分别得到无色油状的2-叠氮乙胺和3-叠氮丙胺,将其溶解在四氢呋喃中,冷却后加入1.0当量的溴乙酸乙酯,分别得到稳定的酯中间体2-叠氮乙基-N-甘氨酸甲酯2和3-叠氮丙基-N-甘氨酸甲酯3。在4M氢氧化钠中同时进行水解反应,并在将pH调整至8.0-8.5后进行Fmoc保护,分别得到Fmoc-叠氮乙基-N-甘氨酸2a和Fmoc-叠氮丙基-N-甘氨酸3a(图2B)。通过乙醚研磨,然后在二氯甲烷和己烷中重结晶,得到纯化合物2a和3a,可直接用于固相肽合成。
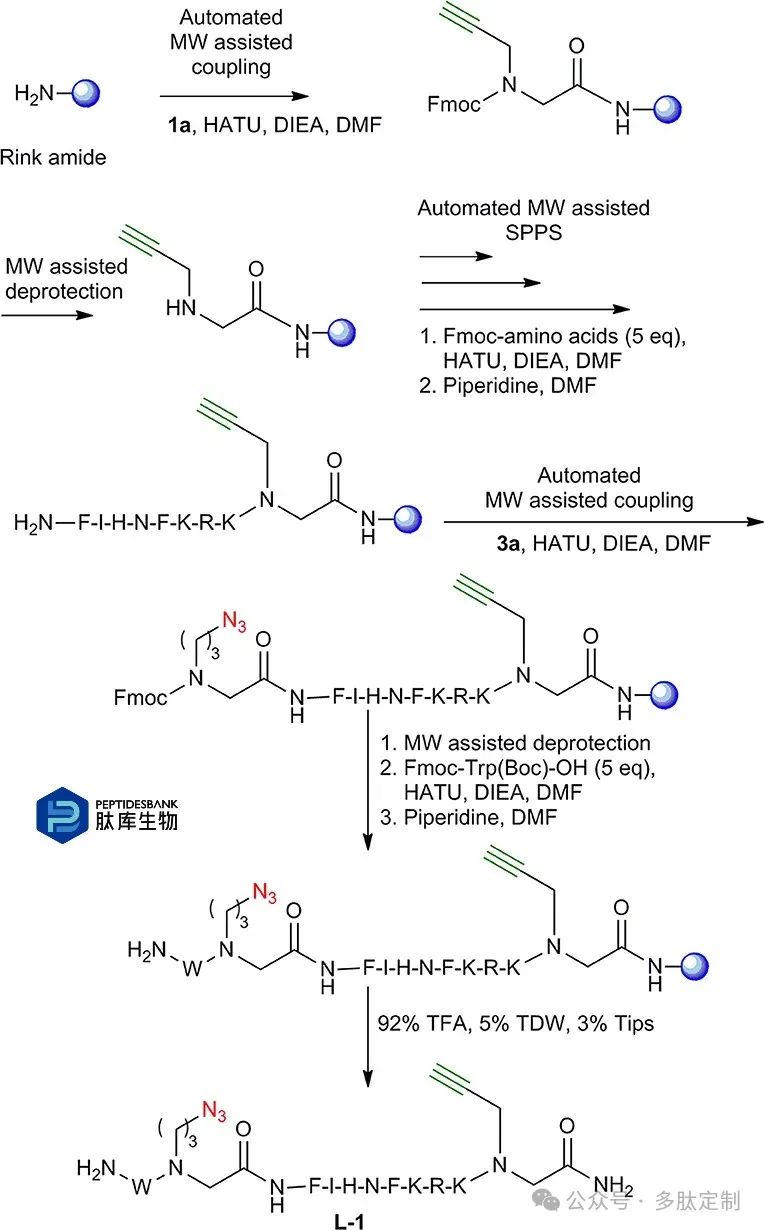
图 3. L-1 的合成。采用微波辅助固相肽合成法合成 L-1 肽 – 肽素杂合物。树脂以蓝色球体表示。
基于 IN181−188 合成肽-肽素杂合物
为了测试新构建模块与固相肽合成(SPPS)的兼容性,我们首先合成了源自IN181−188的线性肽-肽素杂合物。以L-1为例,使用构建模块1a和3a通过分子量辅助的自动化合成方法进行合成(图3)。线性肽L-2的制备采用了类似的策略,只是1a和3a的引入顺序与L-1相反。1a被引入到C端,而3a则在N端的苯丙氨酸和色氨酸之间进行偶联。L-3是使用1a和2a制备的。带有炔基官能团的构建模块1a被偶联在C端,而带有叠氮基官能团的BB2a则在N端。对于L-4的合成,同样使用了1a和2a,但叠氮基构建模块位于C端,而炔基位于N端。在偶联色氨酸之后,去除了Fmoc保护基团,然后通过反相高效液相色谱(RP-HPLC)(图4)、电喷雾电离质谱(ESI-MS)和红外光谱(IR)对线性肽进行切割和分析。
图 4. L-1 肽的环化。 (A) L-1(黑色)、C-1(红色)以及两者混合物(蓝色)的反相高效液相色谱图重叠。L-1 和 C-1 肽从 C18 柱中洗脱的时间不同。 (B) L-1 肽的红外光谱分析显示在 2104 cm−1 处存在叠氮峰,而 C-1 的红外光谱中则没有。
开发肽-肽素杂合物的环化方案
为了优化肽-肽素杂合物的环化方案,再次按照上述方法合成了线性肽L-1,并测试了不同条件下的树脂上微波辅助环化。将CuBr、抗坏血酸钠和二异丙基乙胺的DMF溶液加入带有线性肽L-1的树脂中,在不同温度、反应时间和溶剂系统下进行环化反应。在80°C时,大约15分钟内环化反应完成,通过反相高效液相色谱(RP-HPLC)监测。然而,观察到了几种未鉴定的杂质,总收率不理想。在40°C时,反应时间超过1小时。在60°C下微波辐射20分钟,功率为25瓦的条件下,获得了最高的收率(图5)。RP-HPLC表明,在这些条件下进行环化反应,线性肽L-1转化为环化产物C-1的转化率大于95%。
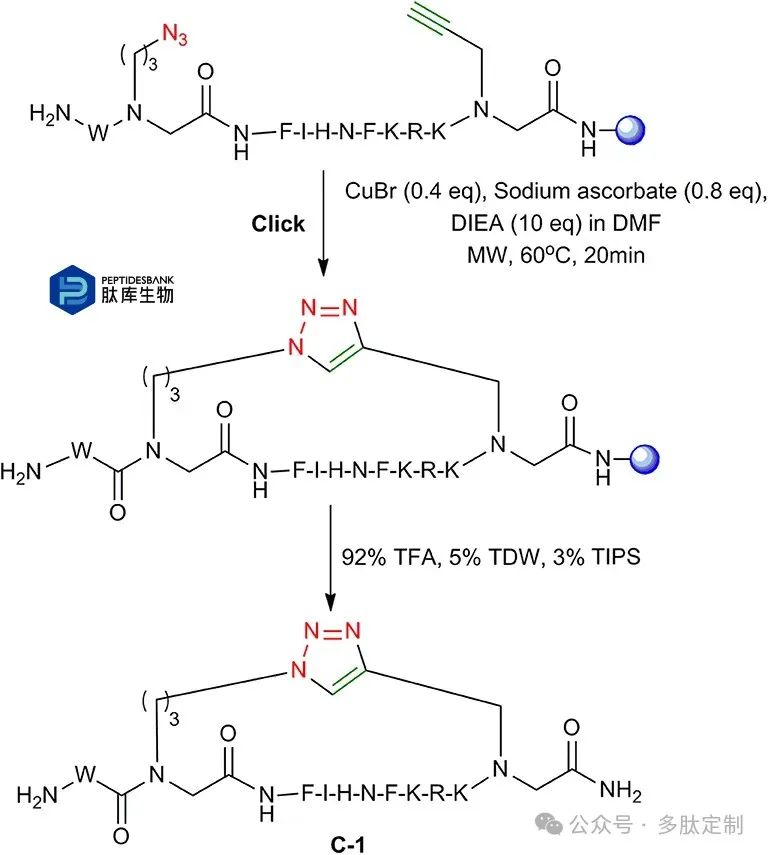
图 5. 固相点击环化。在微波条件下对肽 – 肽素杂合物进行固相点击环化。树脂以蓝色球体表示。
通过反相高效液相色谱(RP-HPLC)对环肽-肽素杂合物的形成进行了监测(图4A)。所有环肽从RP-HPLC柱中洗脱的保留时间均低于其线性类似物,并通过红外光谱(IR)(图4B)和电喷雾电离质谱(ESI-MS)进行了表征。红外光谱分析显示在2000-−1处有一个峰,这对应于L-1的叠氮基团。此峰在C-1肽的红外光谱中未被检测到,表明环化成功(图4B)。
在对模型环肽C-1的环化条件进行优化之后,另外三种环肽C-2、C-3和C-4由其线性母肽在固相支持上合成,然后进行切割。最终环状产物的结构和序列根据叠氮化物和炔烃BB(1a、2a或3a)插入肽链的位置预先确定(图6)。
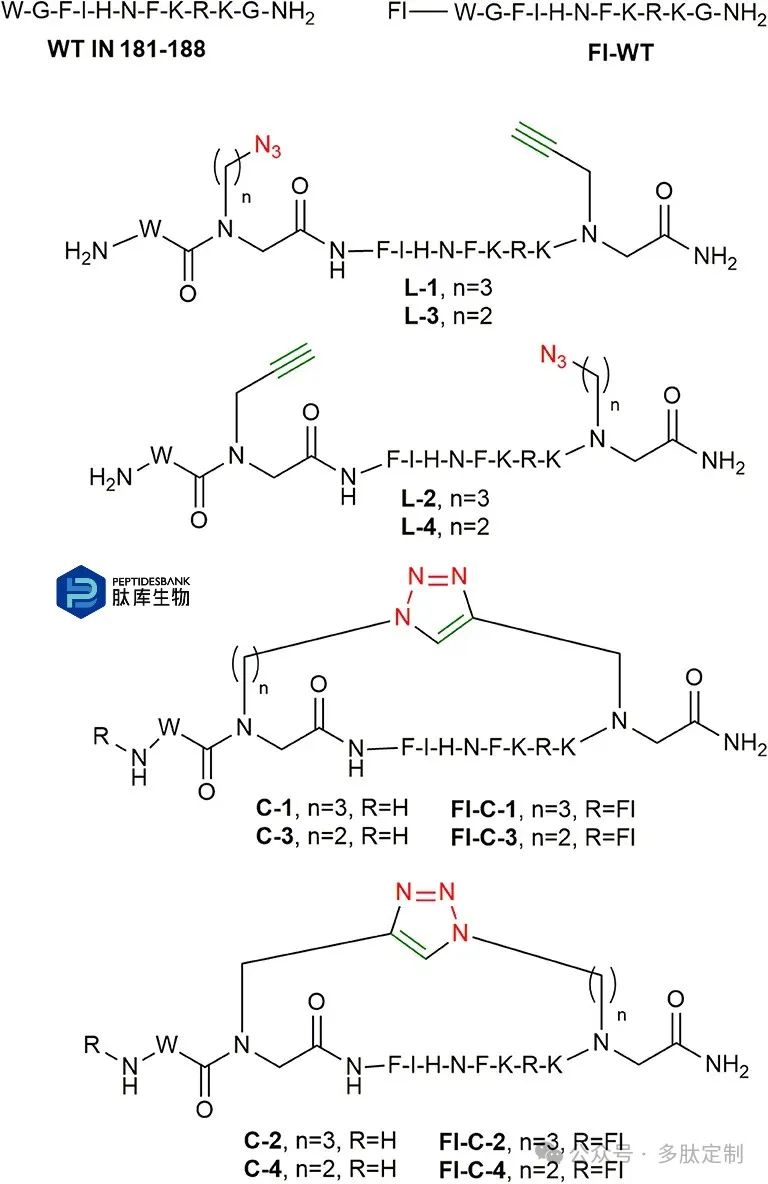
图 6. 本研究中合成的肽。
荧光标记环肽的合成
在优化了线性肽 L-1 至 L-4 的自动化合成策略以及环化得到 C-1 至 C-4 的方法之后,按照所述步骤在固相支持物上将荧光素连接到环肽的 N 端(图 7)。通过高效液相色谱法(HPLC)对荧光素标记的环肽 Fl-C-1 至 Fl-C-4 进行切割和纯化,并通过质谱(MS)进行表征。
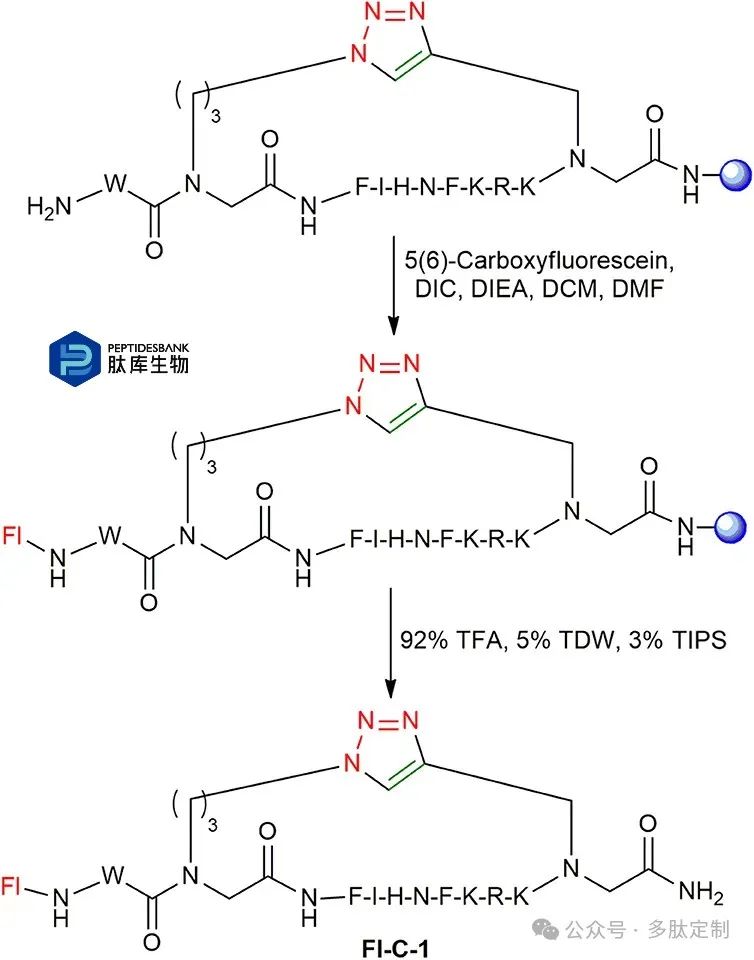
图 7. C-1的荧光素标记。树脂显示为蓝色球体。
为了测试IN181−188环肽-肽素杂合物的活性,我们首先按照先前所述表达并纯化了IN1−/C250S。之所以使用双突变体蛋白,是因为它与野生型具有相似的活性,但溶解度更高。通过荧光各向异性法评估了荧光素标记的环肽Fl-C-1-4与IN的结合情况,并将其与荧光素标记的线性IN181−188肽(WT)Fl-−188的结合情况进行了比较。所有环肽的希尔系数约为4,表明这些肽与四聚体IN的结合具有协同性。IN与所有四种环肽结合的Kd值与IN与Fl-−188结合的Kd值相似,这表明尽管环肽的构象比WT更受限制且环化带来了熵损失,但它们仍保留了对目标蛋白的亲和力(图8,表1)。
图 8. IN181−188 肽与 IN 的荧光各向异性结合研究。展示了线性 Fl-WT(黑色)、Fl-C-1(绿色)、Fl-C-2(蓝色)、Fl-C-3(红色)和 Fl-C-4(橙色)与 IN 结合的曲线及拟合情况。
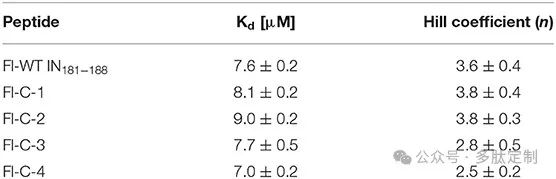
表 1. IN181−188 环肽与整合酶(IN)结合的测得解离常数和希尔系数。
IN 聚合对肽的影响
为了研究 IN181−188 肽对 IN 聚合的影响,我们在有和没有线性野生型肽以及环状 C-1 肽的情况下,对不同浓度的 IN 进行了 AUC 沉降速度实验。较高的 IN 浓度导致在较大的 S 值处出现峰值,表明形成了较大的寡聚体。野生型肽稳定了对应 S 值约为 1.5 的较小寡聚体,而环状肽 C-1 将 IN 的沉降系数从 1.5 到 2 之间移至更高的 2.5,表明形成了高阶寡聚体/聚集体。
一种制备环肽-肽素杂合物的新合成方法
这里开发的策略非常有效,能在2-3小时内完成环化,且在肽的指定位置形成环肽-肽素杂合物。与标准的自动化多肽固相合成相比,这里唯一的额外步骤是在线性肽合成完成后,在指定位置带有叠氮基和炔基的BB上进行点击反应。将点击溶液加入树脂中,并在60°C下额外照射20分钟,即可得到环肽-肽素杂合物C-1-4。该方法不仅减少了步骤,还加快了环肽合成本身的速度。仅需几天时间就能合成目标肽的环状类似物库。
线性肽L-1至L-4的合成采用全自动方法,使用标准的微波辅助固相肽合成技术完成。N-烷基化氨基酸空间位阻较大,通常难以引入或与后续氨基酸偶联。通常采用高活性试剂和高温来克服这些困难。然而,许多此类试剂,如使用三光气作为活化剂,与自动化合成方法不兼容。这阻碍了合成的自动化,使得包含难以偶联步骤的肽库的生产变得非常繁琐。微波辅助固相肽合成技术利用标准协议且可实现自动化,已被用于克服因空间位阻或聚集导致的难以偶联的步骤。然而,在许多情况下,微波程序无法优化,这些步骤仍需手动完成。通过采用微波辅助的自动化固相肽合成方法,加快了L-1至L-4肽库的合成速度。由于自动化微波辅助方法通常采用高温作为标准操作条件,因此在合成过程中也采用了相同的条件来克服与空间位阻较大的N-烷基化构建单元1a、2a和3a进行偶联时的困难步骤。该策略的优势在于无需手动进行任何偶联步骤,整个生成环肽的过程可自动进行且中间无需中断。在此过程中,仅手动进行了环化和裂解步骤。同样的策略也被用于加快C-1-4和Fl-C-1-4库的合成。
选择依靠点击反应来制备库具有诸多优势。首先,经炔基和叠氮基功能化的甘氨酸BB与所有标准固相肽合成(SPPS)方案兼容。其次,标准保护基团和连接子完全不受点击反应条件的影响,因此可以在完全保护的前环肽仍固定在树脂上时进行环化。这种在树脂上进行环化的策略还避免了在溶液相点击环化中更易发生的二聚化和低聚化问题。第三,三唑基团和整个环肽对标准裂解条件稳定。通过改变引入肽序列中构建单元的顺序即可制备具有相反和不同结构的类似物,这与诸如酰胺、脲、二硫键等其他环化方法相比具有许多优势。在其他环化方法中,在肽组装之前和环化步骤之前对保护基团进行操作是一个主要缺点。选择点击化学被证明与整个过程兼容,因为在环化之前无需进行额外的脱保护或裂解步骤。微波辅助环化的优化方案是一项额外的改进,有助于使环肽 – 肽素杂合物更容易获得。它使我们能够快速高效地合成一系列不同的环肽。此外,它克服了使用其他方法进行环化时所需的繁琐的保护基团操作。
环肽对胰蛋白酶消化的稳定性优于野生型
环肽和 N-烷基化肽-肽素杂合物对酶降解的稳定性是采用这种策略所能获得的最重要优势之一,这对改善药理特性至关重要。我们的胰蛋白酶消化研究表明,无论是前环肽还是环肽杂合物,其对酶水解的稳定性都显著高于野生型肽。环肽-肽素杂合物的几种分子和结构特征共同提供了这种更高的稳定性。文库中的环肽具有非天然侧链,两个主链酰胺被烷基化,并且通过酶不自然识别的键进行环化。这些特征中的每一个都可以单独提高稳定性,但它们的组合可以进一步增强稳定性。我们的结果表明,N-烷基化本身(如 L-2 所示)对稳定性有显著改善,与野生型相比,其半衰期显著延长。这种环化作用还具有进一步的优势,因为三唑桥即使在水解后也能防止肽分解成更短的片段,因为酶无法识别它。
在 IN181−188 衍生环肽存在时 IN 的活性
环肽与IN的结合亲和力与线性亲本WT肽相似。在7.8μM时,C-1和C-2对IN的催化活性的刺激程度大于WT肽所引起的刺激。在高肽浓度下,所有四种环肽都刺激了IN的聚集,这已通过AUC结果得到证实。WT和环肽与IN的结合亲和力相同。在低浓度下,WT和环肽刺激IN活性,正如先前对线性INS肽所描述的那样。C-1和C-2对IN的激活程度大于C-3和C-4。在较高肽浓度下,WT刺激IN活性,而环肽则诱导IN聚集,导致活性丧失。我们得出结论,在低浓度下使用环肽是最佳选择——结合了亲和力、活性和稳定性,同时避免了聚集。
总之,采用易于合成的 Fmoc 保护的肽素构建模块,结合自动化微波辅助固相肽合成和微波辅助树脂上正交环化策略,能够快速获得环肽 – 肽素杂合物。这种方法在极短时间内提供了四种环肽 – 肽素杂合物,且与现有策略相比无需额外步骤。我们的方法对于基于肽的药物发现研究很有用,因为这类研究需要一种快速合成稳定且有效的候选药物库的方法。
材料与方法
肽合成及树脂上环化
所有肽均在微波辅助肽合成仪(CEM)上使用标准Fmoc-SPPS化学法在Rink胺树脂上合成,以DIC/氧杂苯并三唑作为偶联试剂。叠氮基官能化构建单元2a、3a与炔基官能化构建单元1a的偶联在75°C的微波条件下于二甲基甲酰胺(DMF)中使用HATU和DIEA的混合物进行5分钟。在整个合成过程中,使用20%的哌啶在DMF中于90°C下进行3分钟以去除Fmoc保护基。点击环化溶液由0.4当量的CuBr、0.8当量的抗坏血酸钠和10当量的DIEA在DMF中溶解而成,并加入到带有线性肽L-1-4的树脂中。环化在60°C下以25W的功率微波照射20分钟进行(图5)。线性和环肽的裂解使用92%的三氟乙酸、5%的三异丙基硅烷和3%的水的混合物进行。裂解后的肽在Merck-上使用反相C18制备柱通过10%至50%乙腈(ACN)在三异丙基硅烷中的梯度洗脱进行纯化。采用ESI质谱分析和高效液相色谱法对肽的纯度和身份进行鉴定。所有肽均经冷冻干燥处理,并在-20°C下保存。荧光素偶联按文献(Weber等人,1998年)所述进行。当使用紫外分光光度法进行浓度测定时,在肽的N端添加一个色氨酸残基。
将环酰胺树脂置于合成仪中,在75°C的微波条件下,用N,N-二异丙基乙胺(DIEA)和1-(3-二甲氨基丙基)-3-乙基碳二亚胺六氟磷酸盐(HATU)的混合物在二甲基甲酰胺(DMF)中与叠氮基官能化构建单元3a进行偶联反应,反应时间为5分钟。在整个合成过程中,使用20%的哌啶在DMF中于90°C下进行3分钟的Fmoc去保护。接下来的八个氨基酸通过标准的微波辅助固相肽合成(SPPS)协议以自动化方式引入。在苯丙氨酸偶联后,使用与3a相同的HATU和DIEA活化混合物,在相同的微波条件下偶联炔基官能化构建单元1a。最后偶联末端的Fmoc色氨酸,从而得到L-1线性肽素-肽杂合物(图3)。
参考文献:doi.org/10.3389/fchem.2020.00405
加入IP合伙人(站长加盟) | 全面包装你的品牌,搭建一个全自动交付的网赚资源独立站 | 晴天实测8个月运营已稳定月入3W+
限时特惠:本站每日持续更新海量内部创业教程,一年会员只需98元,全站资源免费无限制下载点击查看会员权益
站长微信: qtw123cn



